


For decades, Corrective and Preventive Action (CAPA) has held a contentious position in the life sciences industry. Ask any Quality Director about their relationship with CAPA, and you will likely hear a mix of frustration and resignation.
It is simultaneously the most powerful tool for continuous improvement and the single most common source of regulatory findings. FDA Warning Letters cite inadequate CAPA procedures more frequently than almost any other deficiency, yet organizations continue to treat it as a paperwork burden to be endured rather than a strategic asset to be leveraged.
This "check-the-box" mentality is a liability.
In an environment where the FDA is harmonizing with international standards through the Quality Management System Regulation (QMSR), the old ways of managing quality events are no longer sufficient. The shift from the specific prescriptive requirements of 21 CFR Part 820 to the risk-based approach of ISO 13485 represents a fundamental change in how regulators view quality.
It is no longer enough to simply close tickets. You must demonstrate that your quality system is "anti-fragile," capable not just of fixing problems but of predicting and preventing them before they impact the patient.
The following guide explores how to transform your CAPA process from a reactive burden into a proactive engine for operational excellence. We will examine the regulatory shifts driving this change, dissect a best-practice workflow that withstands scrutiny, and discuss how modern technology can automate the heavy lifting of compliance.

The Regulatory Landscape: What "CAPA Quality" Means Now
The most significant shift in the quality landscape is the FDA’s transition to the QMSR, which incorporates ISO 13485:2016 by reference. For years, US manufacturers operated primarily under 21 CFR Part 820, while international companies adhered to ISO standards. The harmonization removes this duality, but it also raises the bar for how companies approach CAPA.
Under the old QSR framework, there was often a heavy emphasis on documentation for documentation's sake. The QMSR, aligned with ISO 13485 Clauses 8.5.2 (Corrective Action) and 8.5.3 (Preventive Action), places a much stronger emphasis on a risk-based approach. This means the rigor of your investigation and the speed of your response must be commensurate with the risk the issue poses to the patient or product quality.
There is also a critical distinction in terminology that often trips up legacy quality teams. "Preventive Action" in the ISO sense is not merely fixing a broken process so it does not break again. That is still part of Corrective Action (preventing recurrence). True Preventive Action is about identifying potential non-conformances that have not yet occurred and taking steps to ensure they never do.
This requires a shift from reactive data management (logging failures) to proactive data analysis (spotting trends). If your CAPA log is 95% Corrective and only 5% Preventive, your system is stuck in firefighting mode.
Regulators are also looking for "closed-loop" quality. They want to see that a complaint received in the post-market phase feeds back into the risk management file, which in turn triggers a CAPA, which then updates the design controls or manufacturing SOPs. If these systems are siloed, you cannot effectively demonstrate this closed loop.
The 7-Step "Best Practice" CAPA Workflow
To move beyond mere compliance, organizations need a workflow that is rigorous yet flexible enough to handle different levels of risk. A robust CAPA process generally follows seven distinct phases.
1. Identification and Risk Assessment
The first step is often where the most damage is done to a quality system. Not every non-conformance, deviation, or complaint warrants a CAPA. If you open a CAPA for every minor documentation error, you will create "Death by CAPA"—a backlog so massive that your quality team cannot focus on the critical issues that actually threaten patient safety.
You must implement a gateway step. When an issue is identified, it should be triaged based on risk.
- Is this a systemic issue?
- Is patient safety at risk?
- Has this happened before?
If the answer is no, a simple non-conformance report (NCR) or immediate correction might suffice. Only systemic, high-risk, or recurring issues should escalate to a full CAPA. This risk-based filtering is explicitly supported by ISO 13485 and helps keep your system lean and effective.
2. Immediate Correction (Containment)
Before you can fix the long-term problem, you must stop the bleeding. Containment involves immediate actions to secure the product and protect the patient. This might mean quarantining a lot of raw materials, issuing a field safety alert, or halting a production line.
It is vital to document this distinct from the root cause fix. Auditors often find companies confusing "Correction" (fixing the immediate mess) with "Corrective Action" (fixing the cause). For example, if a machine leaks oil onto a product, cleaning the product is the correction. Replacing the seal is the corrective action. Both must be documented, but they serve different purposes.
3. Root Cause Analysis (RCA)
This is the heart of the CAPA. If you get this wrong, the problem will return. A common failure mode in life sciences is identifying "Human Error" as the root cause. In modern quality philosophy, human error is a symptom, not a cause. If an operator pressed the wrong button, the root cause is not the operator; it is the process that allowed the wrong button to be pressed, the lack of safeguards, or the confusing labeling.
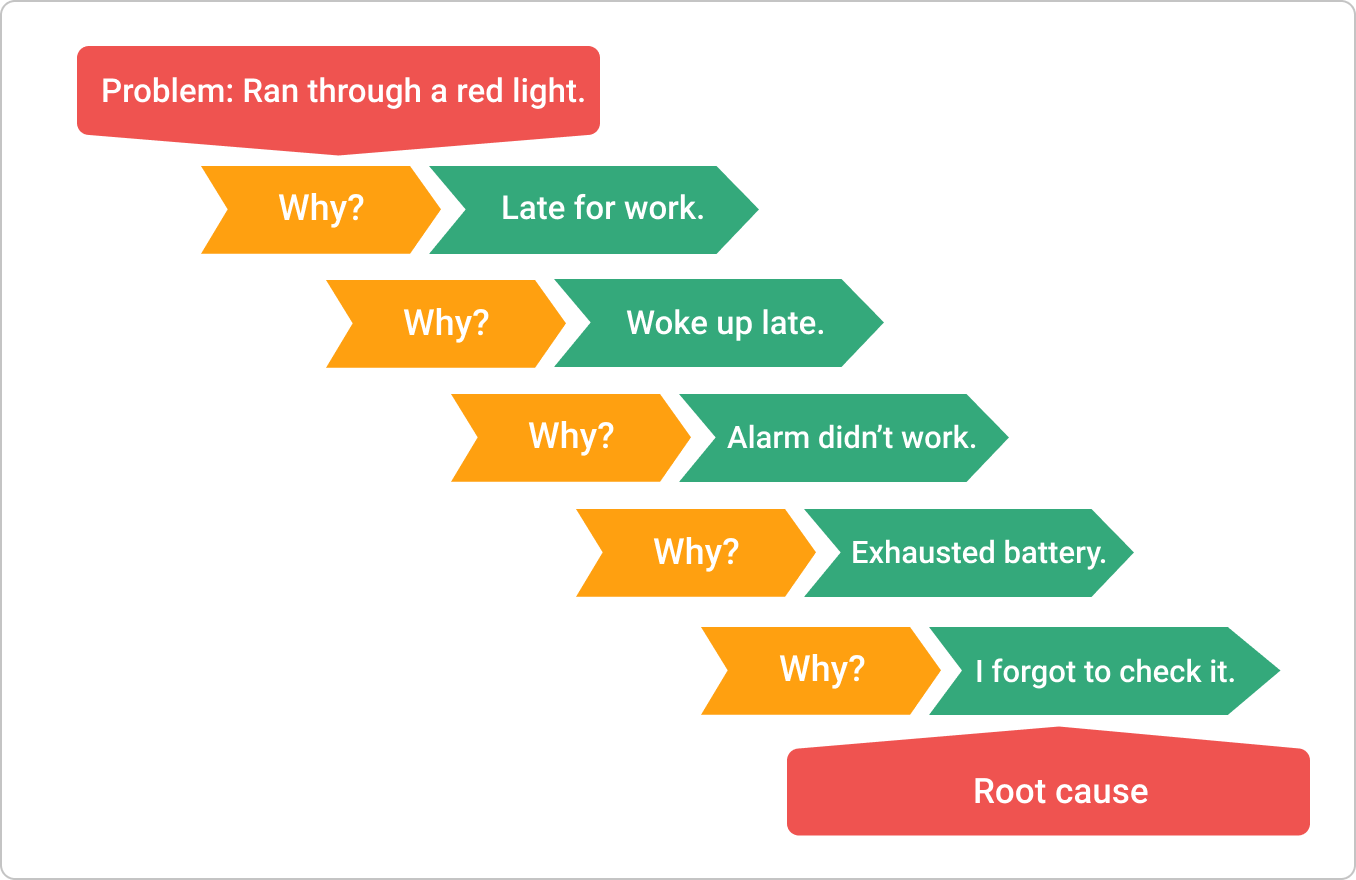
To dig deeper, teams should utilize structured tools like the Fishbone (Ishikawa) diagram or the 5 Whys technique. The Fishbone diagram helps you categorize potential causes into Equipment, Process, People, Materials, Environment, and Management, ensuring you don't develop tunnel vision.

The 5 Whys force you to drill down past the obvious. By the time you ask "Why?" the fifth time, you usually uncover the systemic gap that allowed the failure to occur.
4. Action Plan Planning
Once the root cause is identified, you must develop a plan to eliminate it. This plan should be specific, measurable, and time-bound. It is not enough to say "Update SOP." You must specify which SOP, who will update it, who will review it, and when it will be trained on.
This is also the stage to assess the impact of the change. Will changing this manufacturing process affect the device's validation status? Do we need to re-validate? A good change control process is inextricably linked to CAPA. You cannot effectively plan a corrective action without understanding the regulatory and validation ripples that action will create.
5. Implementation
Execution is where good intentions often fall apart. Implementation involves carrying out the action plan: rewriting the procedure, installing the new fixture, or switching suppliers.
Crucially, implementation must include training. However, "read and understand" training is rarely sufficient for a CAPA. If the issue was complex enough to warrant a CAPA, the training should likely be effectiveness-based, such as a quiz or an observed demonstration of competence. This ensures that the change has actually been absorbed by the workforce.
6. Verification of Effectiveness (VoE)
This is the step most frequently cited in warning letters. Companies often close a CAPA the moment the SOP is updated. This is incorrect. You have only verified that the action was taken, not that the action was effective.
Verification of Effectiveness requires a waiting period. You must let the process run for a statistically significant amount of time or number of batches to see if the issue recurs. For example, if you had a recurring contamination issue, you might need to monitor the next 20 batches over three months before you can confidently say the CAPA was effective. If the problem returns during this period, the CAPA must be reopened, and the investigation restarted.
7. Management Review
Finally, CAPA data must be visible to leadership. ISO 13485 and QMSR require management with executive responsibility to review the quality system's effectiveness.
They don't need to read every CAPA, but they need to see the trends.
- Are CAPAs taking longer to close?
- Are we seeing a spike in supplier-related CAPAs?
This high-level view allows leadership to allocate resources where they are needed most, transforming Quality from a back-office function into a boardroom priority.
The "Quality" in CAPA: Measuring Effectiveness
How do you know if your CAPA system is healthy?
Most organizations rely on lagging indicators, such as the number of open CAPAs or the average cycle time (days to close). While these metrics are useful for resource planning, they don't tell you much about quality. You could close a CAPA in three days by doing a poor investigation; your cycle time would look great, but your quality would suffer.
To truly measure CAPA quality, you need leading indicators and effectiveness metrics. One of the most telling metrics is the rate of "Effectiveness Check Failures." If you frequently get to the VoE stage and find the problem is still there, your Root Cause Analysis process is likely weak. You are fixing symptoms, not causes.
Another critical metric is the "Recurrence Rate." If you close a CAPA and the same issue pops up six months later, your preventive measures were insufficient. High-performing quality teams track these recurrence rates ruthlessly. They also look at "CAPAs generated from internal audits vs. external complaints."
A healthy system finds its own problems (internal) before the market finds them (external). If 90% of your CAPAs come from customer complaints, you are using your customers as your quality control department.
Audit readiness is the ultimate test of your CAPA quality. When an auditor picks up a CAPA file, the "story" should be self-evident. They should be able to read the description, the investigation, the action, and the verification, and understand exactly what happened without asking a single question. If the file is a mess of disjointed emails and vague references, you are inviting further scrutiny. A clean, standalone narrative is your best defense.
Technology and the Future of CAPA
Managing a modern CAPA workflow on paper or in disconnected spreadsheets is a recipe for disaster. The administrative burden of manually tracking links between complaints, investigations, and change controls is simply too high, and the risk of human error is significant.
This is where the modern CAPA management software, most notably your eQMS (Electronic Quality Management System), becomes essential.
Legacy eQMS platforms were often rigid, on-premise behemoths that required months of configuration. The new wave of cloud-native tools has changed this dynamic. Platforms like Kivo have reimagined the QMS as a collaborative, unified workspace rather than just a database of records.
The primary advantage of a unified system like Kivo's QMS is the destruction of data silos. In a manual system, if a CAPA requires a change to a clinical protocol, the Quality team has to email the Clinical team, who then updates a document in a different folder.
In a unified platform like Kivo, the Quality, Clinical, and Regulatory modules share the same document backbone. You can link a CAPA directly to the SOP it affects. When that SOP is updated, the system automatically triggers the required training for all relevant employees.
Furthermore, technology enables real-time collaboration. Instead of routing a paper folder or a Word document via email for approval, teams can collaborate on the RCA and action plan directly in the browser, with a full audit trail of who contributed what and when. This speeds up cycle times significantly.
Common Pitfalls and How to Avoid Them
Even with the best tools, process failures can undermine CAPA quality. There are three specific traps that life sciences companies fall into repeatedly.
The first is the "Death by CAPA" phenomenon mentioned earlier. This usually happens when an organization tries to be "too perfect" or misunderstands the regulations. They open a CAPA for a typo in a logbook. Over time, the backlog grows to hundreds of open items. The quality team becomes overwhelmed, and critical investigations are rushed just to get the numbers down. The solution is a rigorous, risk-based filter at the intake stage. Use Non-Conformance Reports (NCRs) for minor issues and save CAPA for the systemic problems.
The second pitfall is treating symptoms instead of causes. This often manifests as the "Retraining Trap." A deviation occurs, and the CAPA conclusion is "Operator was retrained." While training is often part of the solution, it is rarely the entire solution. If a process allows an operator to make a mistake easily, the process is flawed. You can retrain the operator, but the next new hire will make the same mistake. You must engineer the error out of the process (poka-yoke) or add fail-safes.
The third pitfall is a lack of cross-functional engagement. Quality often ends up owning CAPA alone. They chase down engineers, scientists, or manufacturing leads for information. This leads to weak investigations because the Quality person rarely understands the technical nuance of the machine or the assay as well as the subject matter expert. CAPA must be a team sport. The Quality department owns the process, but the functional departments must own the content of their specific investigations.
Conclusion
CAPA is not just a regulatory requirement; it is the heartbeat of your quality system. It is the mechanism by which your organization learns, adapts, and improves. As the industry transitions to QMSR and embraces a more global, risk-based view of quality, the companies that succeed will be those that view CAPA as a strategic advantage rather than a compliance burden.
The goal is to build a system that is transparent, interconnected, and efficient. By implementing a rigorous 7-step workflow, measuring the right effectiveness metrics, and leveraging unified platforms like Kivo to automate the connections between quality, clinical, and regulatory data, you can significantly reduce your risk profile.
Do not wait for an FDA inspection or an ISO audit to test your system. Start a "CAPA Health Check" today. Pull your last ten closed CAPAs. Read through them. Do they tell a clear story? Did the problem recur? If the narrative is muddy or the issues are repeating, it is time to overhaul your approach. Your patients (and your auditors) are counting on it.