


An Investigational New Drug (IND) application is a mandatory regulatory submission that allows sponsors to legally ship and test experimental drugs or biologics in human clinical trials in the United States.
Key takeaways:
- When needed: Required by law before any human clinical trial can begin
- Timeline: Typically 3-4 months to prepare; FDA has 30 days to review
- Components: Five modules covering administrative, quality, manufacturing, preclinical, and clinical information (~180 documents, ~1,500 pages)
- Success factors: Pre-IND meeting with FDA, complete documentation, and strong safety data
- Cost: No fee for Pre-IND meetings; preparation costs vary widely based on complexity
- Outcome: If no clinical hold is issued within 30 days, trials may begin
Whether you're a first-time sponsor or expanding an existing program, understanding the IND process is crucial for bringing promising treatments to patients safely and efficiently.

Table of Contents
- What is an IND?
- Do I need to submit an IND?
- What are the categories of INDs?
- What are the types of INDs?
- What does the IND include?
- Pre-IND Meeting
- Organizing the IND for the FDA
- My IND is with the FDA - now what?
What is an IND?
Before any experimental drug can be tested in humans in the US, sponsors must navigate the IND application process. This critical regulatory gateway ensures patient safety while enabling promising treatments to advance to clinical trials.
A new Investigational New Drug (IND) Application allows sponsors to legally ship drug or biological product to be used in human clinical trials. Per FDA code 21 CFR 312.20, a sponsor shall submit an IND to conduct a clinical investigation with an investigational new drug and cannot begin a clinical investigation until the IND is in effect.
FDA's primary objectives in reviewing an IND application for Phase 1 trials are to assure the safety and rights of subjects, and in later clinical programs to help assure that the quality of the scientific evaluation of drugs is adequate to permit an evaluation of the drug's effectiveness and safety. FDA's review of Phase 1 submissions will focus on assessing the safety of Phase 1 investigations.
Do I need to submit an IND?
Sponsors under both Commercial and Research INDs need to submit an IND. An IND submission or application to the FDA is required by law (21 CRF 312)1 to start human clinical trials in the US. In addition to the IND being submitted to the FDA, an IRB (Institutional Review Board) or EC (Ethics Committee) must review and approve the study before initiating a trial per 21CFR56 and 21CFR312. The IRB review of the clinical investigation may be conducted in parallel with the FDA review of the IND.
The IND application itself is a collection of approximately 180 documents and datasets. The purpose of the IND is to communicate the sponsor’s intent to start human trials. The sponsor must demonstrate through the pre-clinical and manufacturing documentation:
- The drug is reasonably safe for humans.
- The drug shows some efficacy towards disease or syndrome that warrants commercial development.
Ensuring your IND is complete, compliant, and efficiently reviewed reduces the risk of a clinical hold and possible financial setbacks.
What are the categories of INDs?
The first way that INDs are categorized is based on who is sponsoring them.
Commercial vs Research INDs:
Commercial and Research INDs make up most IND applications. Sponsors (usually a corporate entity) typically submit commercial INDs as the intent is to market the drug/biologic for profit. The research IND or investigator- initiated IND is sponsored by a non-profit institution. Studies run under the Research heading are shorter and may result in publications.
|
IND Submission Volume in 2022 In 2022 FDA received a total of 1865 submissions, including 1124 Commercial and 741 Research IND applications. |
What are the three types of INDs?
The FDA outlines three (3) types of INDs.
- Investigator IND: submitted by a physician who both initiates and conducts the trial, and under whose immediate direction the investigational drug is administered or dispensed. A physician might submit a research IND to propose studying an unapproved drug, or an approved product for a new indication or in a new patient population.
- Emergency Use IND: allows the FDA to authorize use of an experimental drug in an emergency that does not allow time for submission of an IND in accordance with 21CFR , Sec. 312.23 or Sec. 312.20. It is also used for patients who do not meet the criteria of an existing study protocol, or if an approved study protocol does not exist.
- Treatment IND: submitted for experimental drugs showing promise in clinical testing for serious or immediately life-threatening conditions while the final clinical work is conducted, and the FDA review takes place.
Other Special Designations:
Breakthrough Therapy: This designation is for drugs that treat a serious or life-threatening condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on a clinically significant endpoint(s) over available therapies. A sponsor should submit a request for breakthrough therapy designation with the submission of a new IND16. The Food Drug and Cosmetic Act (21 USC 356) states that a request for a breakthrough therapy designation may be made concurrently with, or at any time after, the submission of an application for [the IND].
|
2023 Breakthrough Designation Success Rates For fiscal year 2023, FDA’s two divisions received and granted the following number of requests for Breakthrough designations. CDER Breakthrough Therapy Requests: 60, Granted: 19. CBER: Breakthrough Therapy Requests: 20, Granted: 6. |
Expanded Access (Sometimes referred to as “Compassionate Use”): New drug products outside of clinical trials to treat patients with serious or immediately life-threatening diseases or conditions when there are no comparable or satisfactory alternative treatment options.
What does the IND include?
The IND includes pre-clinical (non-clinical) studies, manufacturing (quality), regulatory (administrative), and clinical documentation and data. Documentation in the form of reports is authored by scientists, chief medical officers, medical writers, data statisticians, chief scientific officers, clinical operations, quality engineers and various other subject matter experts either internal to your organization or sources externally. Raw data are provided in analysis formats (SAS, .xpt), and summaries of that data are represented in tables, listings and/or graphs within the reports.
Broadly the IND application must include information in following modules:
Module 1: Administrative, Regulatory and Regional
Module 1 establishes the regulatory foundation for your IND submission and provides FDA reviewers with essential administrative details about your proposed study.
Key components include:
Cover Letter
- Introduces your submission and highlights key aspects of your development program
- Should be concise (1-2 pages) and written for regulatory reviewers
- Include study objectives, patient population, and any critical safety considerations
- Reference any previous FDA communications or advice received
Form FDA 1571 (Investigational New Drug Application)
- Required cover sheet that legally establishes your IND
- Identifies the sponsor, drug name, indication, and phase of study
- Must be signed by an authorized representative of the sponsor organization
- Serves as a legal commitment to follow FDA regulations
Investigational Drug Brochure (IDB)
- Comprehensive document summarizing all available information about the investigational drug
- Must be updated annually or when significant new safety information becomes available
- Includes pharmacology, toxicology, pharmacokinetics, and any prior human experience
- Should be written for clinical investigators, not patients
General Investigational Plan
- High-level overview of your complete development strategy
- Describes planned studies beyond the immediate IND submission
- Helps FDA understand the broader context and ultimate goals
- Should align with any previous FDA feedback or guidance
Investigational Product Labeling
- Labels for investigational drug containers and packaging
- Must include required safety warnings and storage conditions
- Should clearly identify the product as "investigational"
- Include contact information for reporting adverse events
Correspondence and Meeting Records
- Documentation of all previous FDA communications
- Pre-IND meeting minutes and FDA responses
- Any formal advice letters or guidance received
- Demonstrates collaborative approach with FDA
Module 2: Summaries of Quality and Nonclinical Information
Module 2 provides executive-level summaries that allow FDA reviewers to quickly understand the key quality and safety findings that support human testing.
Key components include:
Quality Overall Summary (QOS)
- High-level summary of manufacturing and quality information from Module 3
- Describes drug substance and drug product characteristics
- Highlights critical quality attributes and control strategies
- Typically 10-20 pages focusing on key quality aspects
Nonclinical Overview
- Executive summary of all preclinical studies from Module 4
- Integrates pharmacology, pharmacokinetics, and toxicology findings
- Provides risk assessment for proposed human studies
- Should clearly justify the safety of the proposed clinical trial
Clinical Summary
- Overview of any previous human experience with the drug
- Literature review of related compounds or drug class
- Integration of nonclinical and clinical data to support trial design
- Risk-benefit assessment for the proposed study
Module 2 summaries are often the first documents FDA reviewers read. They should be well-written, concise, and persuasive, clearly making the case that your investigational product is ready for human testing.
Module 3: Quality Information (Chemistry, Manufacturing, and Controls)
Module 3 demonstrates that your investigational product can be manufactured consistently and safely for clinical use.
This includes drug substance information, like:
General Information
- Nomenclature and structure of the active pharmaceutical ingredient (API)
- Molecular formula, molecular weight, and physicochemical properties
- Description of the drug substance including appearance and solubility
Manufacture
- Detailed description of manufacturing process and controls
- Information about manufacturing facilities and their qualifications
- Batch records and process validation data for clinical supplies
- Supply chain information including raw materials and suppliers
Characterization and Specification
- Analytical methods used to test the drug substance
- Acceptance criteria and justification for specifications
- Stability data supporting proposed storage conditions and shelf life
- Reference standards and analytical method validation
It always includes drug product information, like:
Description and Composition
- Formulation composition including all excipients
- Description of dosage form (tablet, capsule, injection, etc.)
- Rationale for formulation choice and excipient selection
Pharmaceutical Development
- Formulation development studies and rationale
- Compatibility studies between drug substance and excipients
- Container closure system selection and qualification
- Comparison with any reference products or earlier formulations
Manufacture
- Batch formula and manufacturing process description
- In-process controls and testing procedures
- Equipment qualification and cleaning validation
- Batch records for clinical trial supplies
Control of Drug Product
- Analytical procedures for testing finished product
- Specifications and acceptance criteria
- Stability studies and proposed storage conditions
Additional requirements include:
Placebo Information (if applicable)
- Composition and appearance should match active product
- Manufacturing and testing information
- Demonstration that placebo is indistinguishable from active treatment
Environmental Assessment
- Evaluation of environmental impact during manufacture and disposal
- Usually consists of categorical exclusion claim for early-phase studies
- May require more detailed assessment for larger-scale manufacturing
Module 4: Nonclinical Study Reports and Related Information
Module 4 provides the scientific foundation demonstrating that your investigational product is reasonably safe for initial human testing.
You'll need to include information about your pharmacology studies:
Primary Pharmacodynamics
- Studies demonstrating the drug's intended pharmacological effects
- Mechanism of action studies in relevant biological systems
- Dose-response relationships in animal models
- Comparison with existing therapies or standards of care
Secondary Pharmacodynamics
- Studies examining unintended pharmacological effects
- Safety pharmacology studies (cardiovascular, respiratory, central nervous system)
- Evaluation of potential off-target effects
- Assessment of drug interactions with biological systems
Pharmacokinetics and Drug Metabolism
- Absorption, distribution, metabolism, and excretion (ADME) studies
- Species differences in drug metabolism
- Identification of major metabolites and their activity
- Drug-drug interaction potential
You'll also need to include information about your toxicology studies:
Single-Dose Toxicity
- Acute toxicity studies in at least two mammalian species
- Determination of approximate lethal dose and target organs
- Clinical signs, gross pathology, and histopathology findings
- Establishment of starting dose for human studies
Repeat-Dose Toxicity
- Studies of appropriate duration to support proposed clinical trial length
- Identification of target organs and dose-limiting toxicities
- No-observed-adverse-effect-level (NOAEL) determination
- Recovery studies to assess reversibility of effects
Genotoxicity
- Battery of in vitro and in vivo tests for genetic toxicity
- Bacterial reverse mutation assay (Ames test)
- Chromosomal aberration or micronucleus assays
- Follow-up studies if initial tests are positive
Reproductive and Developmental Toxicity (if applicable)
- Required if study population includes women of childbearing potential
- Fertility and early embryonic development studies
- Embryo-fetal development studies
- Pre- and post-natal development studies
Carcinogenicity (usually not required for initial INDs)
- Long-term studies in rodents
- Typically required only for chronic dosing or specific drug classes
- May be deferred until later stages of development
In some cases, special study information might be required as well:
Local Tolerance
- Required for drugs administered by injection
- Evaluation of irritation and tissue damage at injection site
- Studies in relevant animal species using clinical formulation
Immunotoxicity
- Assessment of effects on immune system function
- May be required based on drug class or mechanism of action
- Evaluation of immunosuppression or immunostimulation potential
Phototoxicity and Photosensitization
- Required if drug or metabolites absorb light in relevant wavelength range
- In vitro and in vivo studies to assess light-activated toxicity
Module 5: Clinical Study Protocol and Related Information
Module 5 outlines your clinical trial design and demonstrates your ability to conduct safe and ethical human research.
This starts with your clinical protocol:
Study Objectives and Design
- Clear primary and secondary objectives
- Rationale for study design (randomized, controlled, blinded, etc.)
- Justification for dose selection and dosing regimen
- Statistical considerations including sample size calculation
Study Population
- Detailed inclusion and exclusion criteria
- Justification for patient population selection
- Special considerations for vulnerable populations
- Recruitment strategies and timeline
Safety Monitoring
- Comprehensive adverse event collection and reporting procedures
- Laboratory safety monitoring schedule and parameters
- Dose-limiting toxicity definitions and dose escalation rules
- Data safety monitoring board (DSMB) charter if applicable
Study Procedures
- Detailed visit schedule and procedures
- Pharmacokinetic sampling strategy
- Biomarker collection and analysis plans
- Imaging or special procedures required
Statistical Analysis Plan
- Primary and secondary endpoint analysis methods
- Interim analysis plans and stopping rules
- Missing data handling procedures
- Sample size justification and power calculations
It also includes investigator information:
Form FDA 1572 (Statement of Investigator)
- Signed commitment from each investigator
- Confirms qualifications and agreement to follow protocol
- Lists sub-investigators and study coordinators
- Required for each clinical site
Investigator Curriculum Vitae
- Detailed CV demonstrating relevant clinical research experience
- Board certifications and medical licenses
- Previous experience with similar patient populations or investigational drugs
- Training in Good Clinical Practice (GCP)
Clinical Site Information
- Facility description and qualifications
- Previous FDA inspection history
- Available patient population and recruitment capability
- Laboratory and pharmacy capabilities
Informed consent and ethics information are included here as well:
Informed Consent Form
- Written in language understandable to study participants
- Comprehensive description of study procedures, risks, and benefits
- Clear statement that participation is voluntary
- Contact information for questions and emergencies
Institutional Review Board (IRB) Information
- IRB approval letters for protocol and informed consent
- IRB composition and qualifications
- Continuing review procedures
- Adverse event reporting procedures to IRB
And finally, you'll need to include clinical information, like:
Previous Human Experience
- Summary of any prior clinical trials with the investigational drug
- Literature review of related compounds or drug class
- Analysis of available safety and efficacy data
- Implications for current study design
Risk Management
- Comprehensive risk assessment and mitigation strategies
- Emergency procedures and contact information
- Plans for managing serious adverse events
- Drug accountability and distribution procedures
Plan a Pre-IND Meeting: You won't regret it!
Consider the FDA a valuable resource and partner. Scheduling a pre-IND meeting with the FDA begins a discussion with the project manager. There is no cost to the sponsor to request or have a Pre-IND meeting with the FDA. Plan to request the meeting approximately 60 or more days out from your submission date. Typically, the FDA will respond within 2-3 weeks with a date. Prepare with a briefing package and presentation. The Pre-IND meeting is a type B meeting. The background package (briefing book) should be sent thirty (30) days before the meeting.
Objectives, benefits, and topics to cover at your Pre-IND Meeting
- Understanding proof of concept of the human trial and initiating dialogue.
- Requesting advice for issues related to data needed to support testing in humans.
- Design of non-clinical pharmacology, toxicology, and drug activity studies, including design and potential uses of proposed treatment studies in animal models.
- Data requirements for IND application.
- Initial drug development plans, and regulatory requirements for demonstrating safety and efficacy.17
The meeting is an opportunity to ask questions and request feedback. Establishing the relationship early with your reviewer and project manager will pay dividends in the future.
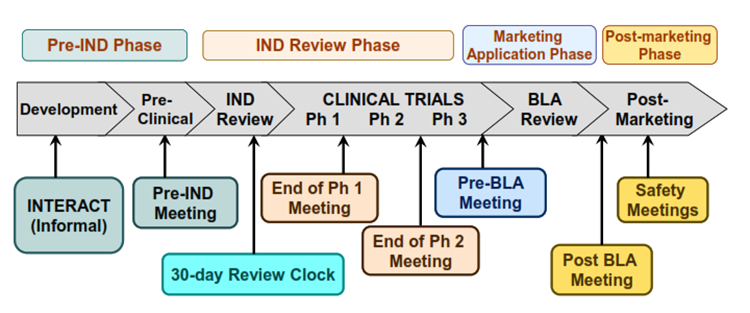
Figure: Opportunities for FDA: Interactions with The Office of Therapeutic Products Website.13
Organizing the IND for the FDA and Submission
Successfully organizing and submitting your IND requires navigating complex formatting requirements, coordinating multiple teams and vendors, and ensuring every document meets FDA standards. The submission process involves transforming scientific data and clinical plans into a structured, regulatory-compliant package that clearly demonstrates your readiness to conduct safe human trials.
Poor organization or missing components can result in clinical holds, costly delays, and damaged relationships with the FDA.
This section outlines the key steps, formatting requirements, and best practices to streamline your submission process and maximize your chances of a successful IND review.
Organizing the IND
FDA accepts INDs in eCTD format. The eCTD format is a globally accepted five (5) module format maintained by the ICH (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use). ICH eCTD is inclusive of Quality, Safety and Efficacy, Administrative/Regional and Clinical documents, and data. Refer to the contents and modules in the sections above for more information.
Module 3 includes Quality, Chemistry, Manufacturing and Controls. Module 4 contains non-clinical reports and data. Clinical documents are filed in Module 5. Summaries are included in Module 2 and Administrative documents in Module 1.
ICH and the FDA provide granular organization down to the document level. For each component of the IND required, there is a corresponding section and document associated. For example, for Pre-clinical Single Dose Toxicology Reports, the section and document number are 4.2.3.1. Single dose toxicity report to be “filed” in the 4.2.3 folder.
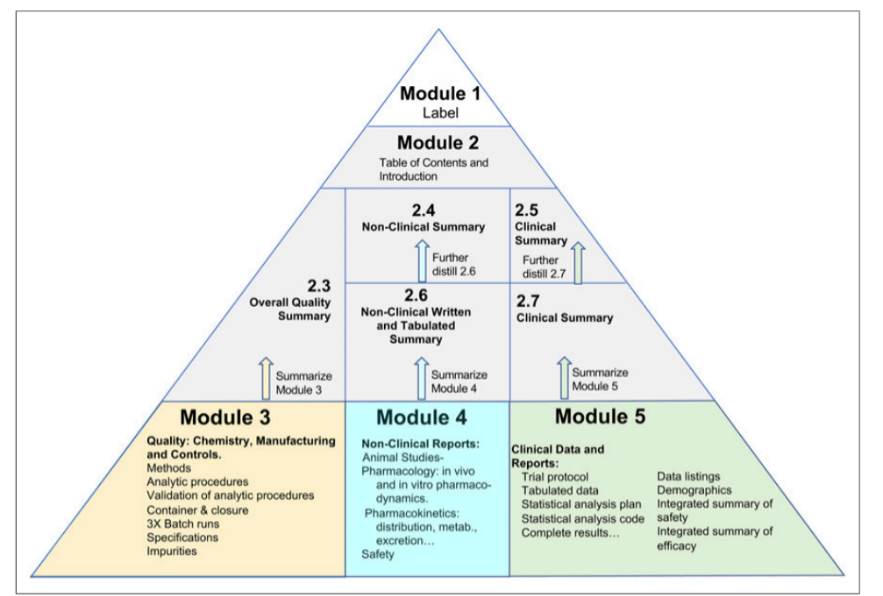
IND Submission
As you consider preparing for the IND submission, aligning resources specific to tasks and timelines will help prioritize and mitigate risk. Resources include the people, systems, and outside vendors to support the business processes.
High level tasks associated with the IND submission include:
- Author, review and approve the content for the ICH 5 modules and sections of the IND. This is typically done inside a RIM system.
- Prepare the PDF content for electronic submissions
- Publish into the eCTD format. Note that eCTD is currently in transition to Version 4.0. Learn more about 4.0 here.
- Dispatch to the FDA
- Archive the submission at the sponsor
FDA and ICH both provide guidance on how the documents and content should be organized for the reviewers. Document templates are available with pre-built sections, tables, and headings to make your life even easier - some templates are publicly available via academic institutions, but most have to be purchased. If you are considering purchasing a template package, be sure to ask how recently it was updated, what macros (if any) they require, and understand how to use the formatting.
Content created for each section of the IND goes through several rigorous processes to ensure completeness and compliance. The first process is authoring creation, review, and approval. Typically, data from animal studies, for example, are compiled in large datasets and analyzed. Content is written from the data and captured in tables, graphs, and listings. Each of these data points must be correlated and checked to ensure accuracy. Once the content is approved by the internal team at the sponsor site, the second process of preparing the documents for regulatory publishing commences. Word documents are converted to PDF, internal hyperlinks and TOCs are built. Regulatory publishing software is required to create the eCTD submission package inclusive of cross document hyperlinks and the XML backbone. The submission package is uploaded to FDA via the ESG gateway.12 The gateway provides a couple of acknowledgements verifying receipt of the submission. Submissions are filed and kept at sponsor sites. If the FDA has any questions, the sponsor needs to quickly refer to the submission and respond.
Below is a flow chart of an example process.
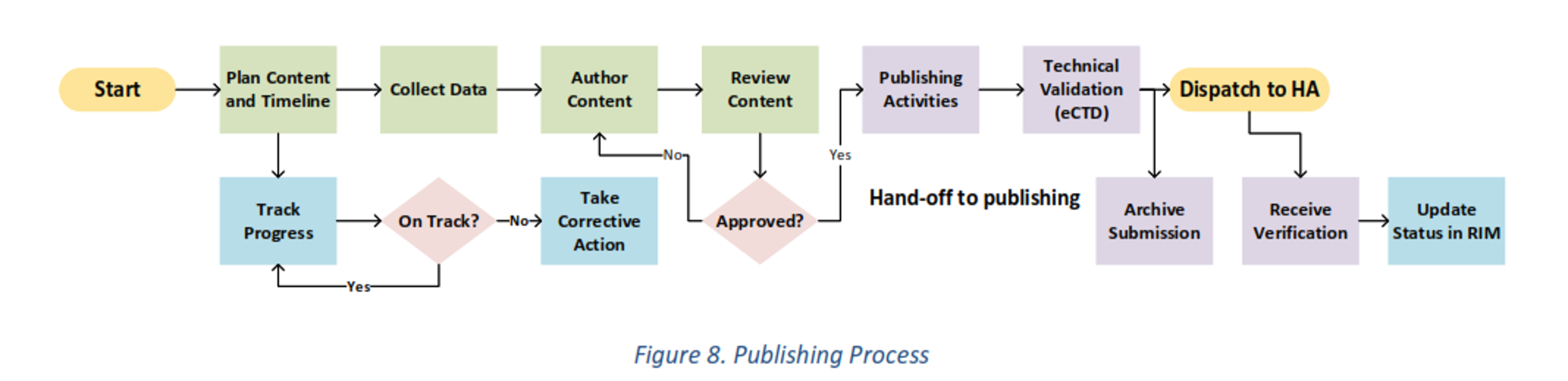
Resources include people, systems, and outside vendors to support the tasks above and the business processes to efficiently meet the submission goals. Examples of resources include:
- CRO vendors for dataset creation and analysis, draft reporting, and writing.
- Collaboration and EDMS solutions for authoring, reviewing and approving content.
- Regulatory Operations publishing for eCTD submission creation, validation and ensuring PDF compliance.
Planning a complete team of people and resources smooths the IND submission tasks and process. Having put the pieces together ahead of time, subject matter experts can focus on the science and messaging in the IND to ensure a successful start of clinical trials.
A word about timelines...
IND timelines vary widely and depend on each sponsors’ program. Drug and biologic research products differ compositionally and as such producing manufacturing protocols do not compare. Outcomes from an FDA meeting could result in performing another non-clinical toxicology study which may add more time. Authoring reports is a cross functional exercise and if a critical subject matter expert is on vacation, the timelines may be delayed. In today's workplace, sponsors work with many vendors who are responsible for portions or all the data, testing and reports. Collaborating closely in a secure, quick, and compliant manner can eliminate delays in retrieving and checking the information. IND projects are usually kicked off within a 3–4-month window of the submission date, though we have certainly seen shorter timelines. Typically, once content is finalized and approved an IND totaling approximately 150-190 documents / around 1500 pages can be published and dispatched to the FDA in 4-6 weeks.
My IND is with the FDA, now what?
The FDA project manager receives the IND and assigns resources. The team has 30 days to review the application in its entirety. FDA reviewers seek concise information supporting the sponsor’s ability to run a human trial safely and provide feedback on efficacy. From the CMC perspective, the agency expects to reduce risk to subjects by defined release testing standards and methods and adherence to Good Manufacturing Guidelines (GMP) for clinical products.
FDA’s resources are inclusive of:
- Clinical
- Non-Clinical pharmacology
- CMC (Chemistry, Manufacturing, and Controls)
- Clinical Pharmacology
- Biostatistics
- Clinical Microbiology
- Microbiology-Sterility
In addition to the subject matters above, the team will cross-reference comparable products previously submitted to FDA and data published in peer reviewed journals.
During the thirty (30) day review period, the reviewer may respond at any time with an RFI (Request for Information). Sponsors must comply and respond in full to the request before moving forward with a trial. If reviewers have many questions or so much information is missing or under safety scrutiny, then the FDA will issue a clinical hold. Some divisions issue a “safe to proceed letter;” otherwise, if no response is received within the 30-day time period, then sponsor may begin the trial.
Best Practices
Applying for a new clinical trial is an exciting milestone for your company. Good planning will ensure the project runs smoothly. If this is your first IND, this is an opportunity to set the tone of your future relationship with the FDA. Taking advantage of the Pre-IND meeting engages the project manager and reviewer about the sponsors program and products.
Best practices suggest sponsors work closely with the CRO, medical writers and internal teams using a collaboration/EDMS system to streamline the handoffs and processes to build efficiencies by delivering content on a rolling basis to the regulatory operations publishing vendor.
Plan ahead, ask your colleagues, read the guidance documents and work with the FDA, and you'll be set up for success!
For more guidance on IND Submissions, or any aspect of the regulatory process, reach out to our team.
How Kivo Facilitates the IND Submission Process
Preparing an IND is a high-stakes milestone. Most teams are juggling hundreds of documents, multiple vendors, and strict timelines. Delays or incomplete submissions can trigger clinical holds, push back investor milestones, and slow your path to patients.
Kivo was built to eliminate these roadblocks. Our unified document management system brings quality, regulatory, and clinical content into one platform, so your team and partners work from a single source of truth. That means:
-
Centralized collaboration: Authors, reviewers, and CRO partners can contribute in real time without version conflicts.
-
eCTD-ready organization: Content is structured to match FDA expectations, reducing the time and stress of publishing.
-
Validation you can trust: Kivo’s GxP-compliant environment is audit-ready and speeds internal approvals.
If your team is approaching its first IND, or scaling submissions across multiple programs, Kivo can help you get there faster and with greater confidence.
👉 Schedule a demo below to see how Kivo can streamline your IND submission process and set your program up for success.

References
- Code of Federal Regulations. 21CFRpt 312- Regulation on when an IND is required.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-D/part-312 - FDA Code 21 CFR 312.23 Content of an IND
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-D/part-312#312.23 - FDA Code 21 CRF 56 Institutional Review
Boards. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-56 - FDA Draft Guidance “Formal Meetings Between the FDA and Sponsors or Applicants of PDUFA Products” (December 2017) https://www.federalregister.gov/documents/2017/12/29/2017-28140/formal-meetings-between-the-food-and-drug-administration-and-sponsors-or-applicants-of-prescription
- FDA Guidance for IND Meetings for Human Drugs and Biologics: Chemistry, Manufacturing, and Controls Information (May 2001) https://www.fda.gov/regulatory-information/search-fda-guidance-documents/ind-meetings-human-drugs-and-biologics-chemistry-manufacturing-and-controls-information
- FDA IND Applications for Clinical Investigations: Chemistry, Manufacturing, and Control (CMC) Information; https://www.fda.gov/drugs/investigational-new-drug-ind-application/ind-applications-clinical-investigations-chemistry-manufacturing-and-control-cmc-information
- FDA: IND Applications for Clinical Investigations: Clinical Protocols; https://www.fda.gov/drugs/investigational-new-drug-ind-application/ind-applications-clinical-investigations-clinical-protocols
- FDA Requesting a Pre-Assigned Application number; https://www.fda.gov/drugs/electronic-regulatory-submission-and-review/requesting-pre-assigned-application-number
- FDA eCTD Resources. https://www.fda.gov/drugs/electronic-regulatory-submission-and-review/ectd-resources
- FDA IND Receipts https://www.fda.gov/drugs/ind-activity/ind-receipts
- FDA Forms and Instructions https://www.fda.gov/drugs/investigational-new-drug-ind-application/ind-forms-and-instructions
- E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6(R1); https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e6r2-good-clinical-practice-integrated-addendum-ich-e6r1
- Milstein, J. Lwin, E., “How to put together an IND application.” Lecture presented at FDA Clinical Investigator Training Course. 2018.
- FDA Gateway Page: https://www.fda.gov/industry/electronic-submissions-gateway; https://www.fda.gov/industry/about-esg/esg-submission-process.
- FDA: Interactions with The Office of Therapeutic Products. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/interactions-office-therapeutic-products
- FDA : Frequently Asked Questions: Breakthrough Therapies. https://www.fda.gov/regulatory-information/food-and-drug-administration-safety-and-innovation-act-fdasia/frequently-asked-questions-breakthrough-therapies
- “Demystifying the Investigational New Drug (IND) Application for Drugs and Biologics” video. US Food and Drug Admin. Bugin, Kevin. Office of New Drugs. CDER. REdI Conference-Spring 2018.