


Revised March 24th, 2025
The transition to eCTD 4.0 is one of the most significant regulatory updates in recent years, with agencies worldwide beginning to adopt this new standard. If you are involved in regulatory affairs, now is the time to ensure your organization is ready for this shift. The U.S. FDA and Japan PMDA are already accepting eCTD 4.0 on a voluntary basis, and mandatory adoption deadlines are approaching. Failure to update your submission processes in time could result in compliance risks, submission rejections, and delayed approvals. Understanding what eCTD 4.0 entails, how to update your processes, and the consequences of non-compliance will help you prepare for a seamless transition.
A Quick History of eCTD
It’s been 20 years since the eCTD XML format was adopted. Over the years, the industry has evolved to push the current eCTD XML standards to handle more types of submissions. Other formats, such as RPS (Regulated Product Submission) created by HL7, have been discussed. RPS includes veterinary, device, tobacco products, and human drugs/biologics. eCTD v. 4.0 is the natural next step from version 3.2.2.
ICH started developing eCTD v. 4.0 in 2010. Implementation guides began surfacing in 2016. In 2015, the ICH endorsed a full package of controlled vocabularies, schemas, transition plans, and various other technical files. Version 4.0 is based on HL7 standards and RPS. The latest version extends XML functionality, allowing document reuse and flexible software updates for regulatory changes.
What’s New in eCTD 4.0?
Key Enhancements:
- Single XML Backbone: M1 regional and M2-5 documentation consolidated into one XML file.
- Improved Document Reuse: Unique Identifiers (UUIDs) allow easier referencing of previously submitted content.
- Advanced Lifecycle Management: New lifecycle operators provide greater control over document updates.
- Controlled Vocabularies (CVs): Standardized metadata terms streamline classification and searchability.
- More Flexible Submission Structure: Less rigid formatting benefits both agencies and sponsors.
| The intent of eCTD v.4.0 is to increase speed, flexibility, and communication in regulatory submissions. |
How Does eCTD 4.0 Affect Me?
Mandatory adoption deadlines are approaching, with most agencies requiring compliance by 2028. Now is the time to educate yourself and your team on the terminology and basic concepts.
For Executives:
- Eliminating costly software upgrades due to simpler DTD updates.
- Reducing submission workload through content reuse.
- Streamlining global submissions.
For Regulatory Affairs & Operations:
- Reducing regulator review time by grouping and prioritizing content.
- Minimizing rework due to inaccuracies in file names.
- Faster submission creation for multi-region applications.
The Benefits of Upgrading to eCTD 4.0
- Submit in the same format across ICH regions.
- Change document granularity while maintaining lifecycle relationships.
- Set document order within a CTD section or assign priority.
- Identify submission content for additional processing (e.g., datasets).
- Reuse previously submitted documents.
- Use controlled vocabularies for standardization.
- Correct attributes and metadata more easily.
- Ensure consistent grouping of documents across ICH regions.
- Uncouple software upgrades from DTD updates.
What Happens If You Don’t Upgrade?
The FDA will mandate eCTD 4.0 for regulatory submissions by 2028. Failure to transition in time could result in:
- Submission delays or rejections due to non-compliance.
- Increased regulatory scrutiny and audit risks.
- Inefficient document management leading to higher costs.
- Lost opportunities for faster approvals in competitive markets.
What Do I Need to Do Differently for eCTD 4.0?
Your regulatory publishing software or outsourcing vendor will handle most technical aspects. However, oversight activities are essential to ensure compliance. New or changed concepts include lifecycle operators, controlled vocabulary options, context of use, and content reuse.
Reuse of Content
Unique Identifiers (UUIDs) allow reuse of content across sequences, regulatory activities, and applications. This reduces the size of new sequences and eliminates confusion over duplicate content.
A key challenge for sponsors is tracking and tagging original content within their internal document management solution (EDMS). Ensuring that links to original documents are maintained is critical for lifecycle management.
New Lifecycle Operators
New lifecycle operators give sponsors greater flexibility in updating submissions:
- Active, Replace, and Suspend (previously New, Replace, Delete).
- Replace Options:
- Replace one with one.
- Replace one with many.
- Replace many with one.
- Maintain lifecycle relationships on all documents.
- Replace Options:
The replace function will track the changes made to the submission contents when one document (referenced by a context of use) is replaced by one, many, or many to one.
FDA guidance provides this example below:
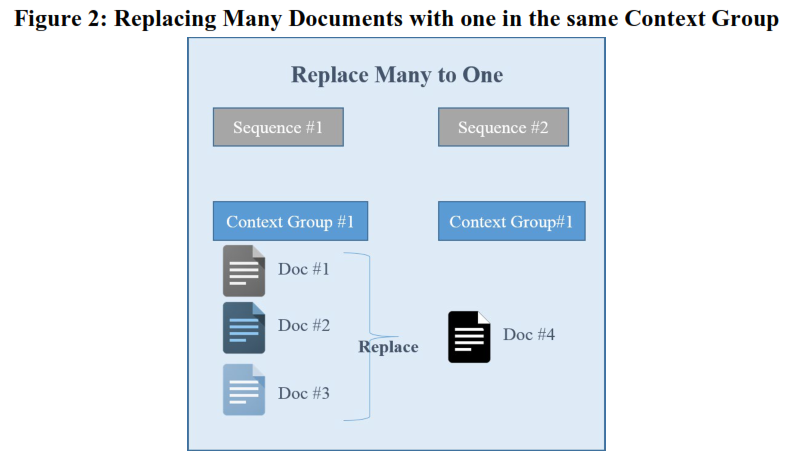
Context of Use
Context of Use (CoU) is similar to the leaf title. It determines the placement of the document in the CTD structure and associated keywords. Content of Use indicates the lifecycle of the document. Combining Content of Use groups, document type, and keyword replace the STF by organizing multiple files relating to a single study in M4 and M5. Additionally, Context of Use allows the advanced lifecycle operations described above to replace one with many and many with one.
Controlled Vocabularies
When combined with the content of use, controlled vocabularies (CVs) further describe and define content within the submission. Five types of established CVs are accepted for eCTD v4.0: ICH, HL7, Regional, sender-defined, and external organizations-ISO Country Codes.
Examples include ICH CV for species: mouse, and rat. US regional CV includes NDA, IND, and ANDA. New CVs introduced for v4.0 provide additional content or replace older values. Examples are Category Event, Document Type, and Study Group Order.
Sponsors define (sender-defined) keywords like the name of a manufacturer (fictional example: ABC CDMO) or the name of the drug. All regions will receive the same message since the keywords are included in the XML packages.
ICH describes in detail the use of both Content of Use and Keyword combinations to place and describe M3 Drug Substance content.

Below is an example of the CMC Amendment mapped to the content of use, keywords, and other required areas of the new structure based on the ICH implementation guidelines. A new contract manufacturer, ABC CDMO, is being added. ABC CDMO manages the creation and distribution of the drug substance, DS.
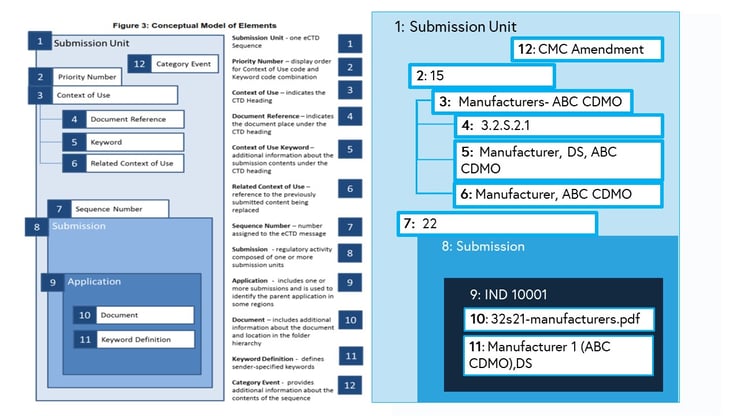
By moving towards a more data-driven approach, sorting and editing the information about the submission becomes a simple process. V4.0 creates a path towards a more flexible submission structure for sponsors and streamlined review for agencies.
Metadata Corrections
Corrections are much easier using keywords. Typos in attributes and keywords are easy fixes, where previously, sponsors were required to submit a new document with the correct file name.
Order and Group Documents in a Section
Using priority numbering, regulatory leaders now control the order of documents in a section. Giving clear priority to content for reviewers expedites reading timelines and creates efficient responses to information. Even better, grouped documents and ordering within a study further organize the M4 and M5 content for the reviewers. Order and Groups replace the STF.
Table of Contents
Gone are the days of the hierarchy. Documents and files are delivered in a flat structure. Within the file name, the Context of Use (CoU) and keywords place the content in the correct location, and keywords provide additional detail to the content of use (CoU).
eCTD Viewer
In the older versions, a style sheet was included. Users could open the .xml file in a web browser and see/navigate a submission as a standalone sequence. With v4.0, this is no longer the case. An eCTD viewing tool will be required, either integrated within an EDMS or publishing solution or standalone.
What stays the same?
Documents: Sponsors will still write documents in the ICH granular format in Word. Conversion to PDF format is still required. Authors continue to utilize the granular ICH CTD documents for each appropriate section of the five (5) modules.
EDMS, Regulatory Operations Publishing, and RIM: Reliance on systems, especially EDMS, will continue to become more important in tracking documents submitted. Regulatory Publishing solutions must adapt to the XML formats and support the various Controlled Vocabularies required, including sponsor-generated CVs. Currently, many EDMS and publishing integrations include the sharing of submission metadata. RIM solutions may encompass both of these solutions and various other functions for tracking, preparing, and reporting on regulatory activities.
How do I prepare for eCTD 4.0?
Japan PMDA and the US FDA are accepting eCTD v4.0 voluntarily. Participating during the voluntary time period is an option for learning with the agency. With the FDA, the process starts with a conversation with a project manager and determining a process. The FDA started accepting production eCTD v4.0 submissions in Q1 2024. It is never too early to start your team on the educational journey!
Key steps and areas to focus:
- Educate yourself and your team on the new metadata and concepts, such as the context of use.
- Build, align, and incorporate a sponsor keyword list into your systems now. For example, use EDMS metadata to drive compliance.
- Keep the new guidance in mind as a requirement when upgrades or new technology solutions are presented.
- Consider participating with a pilot during the voluntary time period.
Lastly, we—technology providers, regulatory agencies, and sponsors—are all on the journey together towards eCTD v4.0. The more we can share each other's experiences and learn, the better the entire industry will be. Attend the conferences, watch the webinars, and read the guidance, and we'll be on our way to harmonization!
References:
- ICH eCTD v4.0 Implementation Package
- ICH M8 technical specification, eCTD v4.0 IWG Question and Answer and Specification Change Request Document
- FDA Electronic Common Technical Document (eCTD) v4.0 Technical Conformance Guide
