


RIM (Regulatory Information Management) refers to the effective and efficient identification, collection, curation, communication, and management of regulatory information for products across the life sciences value chain1. RIM systems encompass business processes from regulatory dossier submission planning and tracking to health authority correspondence and commitment tracking to product registrations to electronic document management. Each of these business process areas help life science companies organize, communicate, manage, and identify their products. Together they create a powerful ecosystem of shared metadata, documents, and other key descriptors to report on progress, areas of improvement and overall performance.
Do I really need a RIM System?
Pharmaceutical and Biotech companies perform RIM activities without a vendor supplied end-to-end RIM solution all the time. The systems are simply more manual. For example, we often meet with emerging life sciences companies using the following configurations:
|
Business Process |
Manual Method |
|
EDMS and Collaboration |
Box, an unstructured SharePoint, or Email |
|
Submission Management |
Outsourced |
|
Health Authority Commitments/Label Tracking/ Registration Tracking |
Asana, Monday, MS Project, EXCEL |
|
Regulatory Intelligence |
See EDMS |
While using the above configuration does get the job done, the systems are disconnected, prone to human error, lack the ability to create auto reports and are not inherently compliant.
🧐 Wondering if it's time to migrate off SharePoint? Check out our article.
For example, during an IND application review, FDA sends a Request for Information (RFI), and the sponsor must respond within a matter of weeks to ensure the start of the trial on time. Examples of Requests for Information from FDA:
- Investigator brochure is misleading, erroneous, or materially incomplete.
- IND application does not contain sufficient information needed to assess the risks to subjects of the proposed studies.
- CMC stability and purity issues.
- Pharmacology and Toxicology Data is difficult to interpret.
Using manual processes, regulatory teams and leaders frequently stress over locating the correct original document in question and coordinating with subject matter experts (SMEs). The SMEs update the content in question and route for review and approval through an unsecure, non-audit trail tracking system. In addition, collating edits to compile a comprehensive response is a nightmare if systems fail by losing data during the review process. Finally, updating the project tracking tasks requires a human to remember to check off. At the end of the response and hopefully a successful review, the trial has started. Now, executive management requests a report on the metrics of how the entire IND project went and how the team “improve” timelines on the next submission.
As you can imagine, most of these companies are looking to modernize, streamline and become compliant with an off-the shelf RIM software solution. The suite of business processes and functions available in a RIM solution solves many of the headaches above.
What does a RIM System do?
The goal of a purpose-built RIM software or system is to streamline and empower people by providing a simple, straightforward mechanism to use and work efficiently with unstructured product data and content for regulatory events and communications.
Features of a typical RIM System:
RIM solutions vary in scope, depth, and breadth. Most will include some basic ingredients to fulfill requirements. Gens and Associates has identified 14 key areas (15 inclusive of medical device). In their yearly report on industry, they further discuss functionality by investment within the size of small, mid, and large life sciences organizations. Maturity of RIM systems deepen as organizations grow and learn from implementing and using the systems. Many firms start with simply finding, identifying and organizing content, developing workflows, and creating operational reporting on metrics in the first years. Later, as the systems and team gain experience, reporting turns to forecasting and resource planning. Additionally, a wider use of integrations with TMF and pharmacovigilance share not only metadata but reduce duplication and errors in version control. These mature companies also trend towards acceptance of global standards like IDMP.
One key area as a starting point or building block is the EDMS (electronic document management system). The EDMS provides the basis for many of the other areas to function and is the foundation platform to grant user permissions, build reports, control security and capture audit trails. As life science companies fall under GCP and GMP guidance, an adequate document compliance system is required by US FDA and EMA for control of documentation regarding clinical trials, regulatory submissions, and manufacturing procedures.
🧐 Wondering how to choose an EDMS? View our guide here.
The RIM capabilities listed below are typically included in a solution depending on the vendor and software provider. The scope to which each is provided may vary. Some vendors choose to provide an end-to-end platform solution. This type of solution will likely encompass many of the areas below in an all-in-one user interface and similar user experience. Other solutions may provide a few core capabilities and integrate with other providers to give customers a full RIM experience. We often see this example with regulatory publishing vendors who integrate with EDMS providers.
|
Key Capabilities of a RIM System: |
|
1) Submission Forecasting and Resource Planning |
|
2) Dossier Management (content plan, distribution, archive) |
|
3) Submission Document Management |
|
4) Electronic Submissions |
|
5) Submission Planning and Tracking |
|
6) Product Registration Management |
|
7) Health Authority Commitment Management |
|
8) Health Authority Interactions (Q&A, correspondence) |
|
9) Regulatory Archive |
|
10) Label Management |
|
11) Reporting, Analytics, Dashboard |
|
12) Data Standards and Governance Management |
|
13) Design History File (Medical Device) |
|
14) Regulatory Intelligence |
|
15) Ad / Promo |
Each of the functional feature areas comes with its own set of workflows and requirements. There are often shared users, metadata, documents and overlap in access.
Why do I need a RIM system?
RIM systems enable connected information to empower regulatory processes. Emerging life science companies to large pharma reap the benefits of a RIM system. Every life sciences organization evaluates their requirements through the lens of budget and goals based on number of drug or biologic products in the pipeline.
For example, an emerging pharma focuses on core operational functionality within RIM. With smaller teams and pipelines early in development often in US and EU markets, EDMS, submission planning/tracking, and health authority commitments are top of mind. Layering correspondence, submission archive and reporting enable history and metrics to emerge. Investments are made operationally on end-to-end processes around new submission requirements like the EU CTA process and CMC change control processes.
All sized life science companies benefit from a RIM solution by:
- Accessing regulatory information in real-time.
- Reducing submission operations complexity.
- Improving consistency of Health Authority interaction, specifically an inspection.
- Better integration of business processes i.e., TMF or Quality
RIM software solutions achieve the benefits above by enabling life science teams in the specific functions below.
- Identifies the product information needed by global regulatory agencies.
- Collects and provides access to Regulatory Intelligence to drive informed decisions.
- Controls the dossier configurations to account for variation in products and regulatory agencies.
- Manages changes and revisions to labeling, advertising and communications documents to reduce errors and delays in approval.
- Generates complaint submission documents, manages changes and revisions.
- Issues and tracks submissions across global agencies.
- Minimizes effort and calendar time to replicate regulatory submissions between products and regulatory agencies.
- Reduces risk and delays by identifying and managing health authority commitments and correspondence.
- Provides clear oversight of original and lifecycle submissions.
- Fulfills compliance requirements of electronic systems, such as 21CFR§11.
- Forecast and resource for regulatory activities via reports.
- Integration, sharing metadata or user access with TMF and Quality systems.
RIM solutions by and large streamline regulatory activities and reduce delays and risk due to manual processes and duplicated documentation. From authoring and collaborating at the EDMS through to a marketing authorization inspection and beyond to post market safety and CMC updates a RIM solution supports the entire development cycle. RIM investment pays for itself sooner rather than later. Typically, teams see a ROI within one to two years.
How to Implement a RIM System
A RIM implementation is really no different than any other software implementation that your team may encounter. The implementation follows most steps as outlined below in the image. Some areas of note are compliance, configuration, roll out of features and 3rd party consulting.
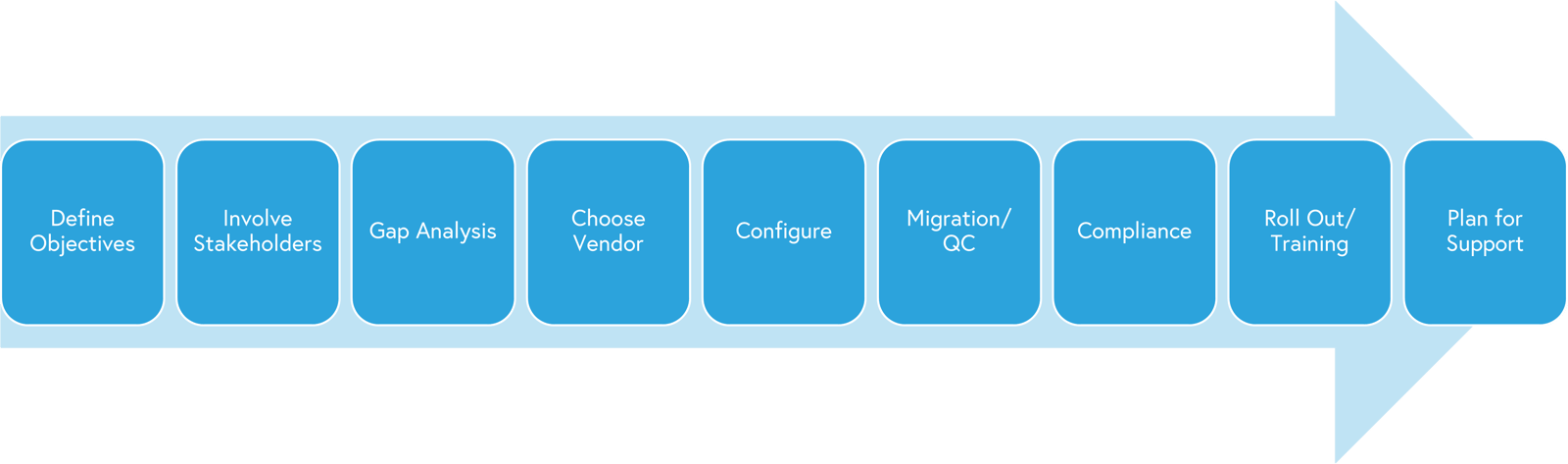
Considerations unique to RIM include regarding the various outside vendors and constellation of contractors given the compliance and security requirements. Considering the CRO, CMO, Medical Writers or other critical contributors early and including in workflows, configuration and data integrity models will save time and headaches later. Try to ensure flexibility for handoffs, auto tracking for quality/audit reporting and availability of user accounts.
Life sciences organizations must follow GCP and GMP compliance for data and computer systems. Most of the software vendors supplying RIM solutions are aware of the requirements, but ultimately it is the sponsor’s responsibility to demonstrate control and documentation. Be sure to ask and receive the validation documentation as part of your deliverables.
Other considerations are choosing to implement RIM functionality to meet the needs and requirements for the organization. For example, Company K is an emerging pharma with one (1) product with an active IND and CTA with two (2) phase 1 trials and one (1) more planned IND to FDA in the next 6 months. Company K’s top requirements are computer systems compliance, control and access of regulatory documents, submission management and health authority correspondence/commitment management. Company K’s best bet may be to explore a rolling feature implementation. Choosing to implement functionality based on top requirements first, followed by secondary requirements later.
Another example is Company Q with three (3) active IND/CTAs in various phases of development, two (2) marketed products and one (1) BLA planned for the end of year with a simultaneous MAA to EU. Company Q’s requirements are more complicated. Company Q will utilize the full suite of RIM functionality from day one. These two (2) companies will have different implementation plans and processes. Additionally, integration points and vendor selection will vary greatly between the two.
In addition to partnering with a software vendor for the RIM software itself, there are several implementation validation, data migration, and data governance consulting firms available on the market for assistance. Depending on the needs and requirements of your project, a 3rd party group may be helpful. Typically, we see this for complicated projects with several integration points and large complex data migration.
Unique RIM Implementation Tips:
- Consider your outside contributors for licensing, workflows, user access, configuration, and security.
- Ensure proper documentation of the computer systems validation.
- Implement features and functionality on a rolling basis as needed.
- Consider a 3rd party consulting firm as a partner if implementation is complicated.
A full discussion of how to manage a RIM implementation is coming soon, stay tuned!
References:
- Balasubramanian V., Mahoney-Jewels S., Brewer-Yizar V., et al., “Regulatory Information Management Whitepaper V2,”, DIA Regulatory Affairs Community Regulatory Information Management (RIM)Working Group. Jan 21, 2019. Accessed May 2, 2023. https://www.diaglobal.org/en/resources/tools-and-downloads#RIM.
- Gens S., Brolund G., Hnat K., Powell S., Yang-Iott K., 2022 World Class Regulatory Information Management Study White Paper. July 2022. Accessed July 31, 2023. https://gens-associates.com/world-class-rim-research/.